PRIME Tutorial
Overview
This clustering tutorial is meant for datasets Molecular Dynamics Trajectory. PRIME assumes a MD trajectory that has a well-sampled ensemble of conformations. The PRIME algorithm predicts the native structure of a protein from simulation or clustering data. These methods perfectly mapped all the structural motifs in the studied systems and required unprecedented linear scaling.
Tutorial
The following tutorial will guide you through the process of determining the native structure of a biomolecule using the PRIME algorithm. If you do not have clustered data. Please refer to other clustering algorithms such as NANI to cluster your data, follow all steps.
1. Clone the MDANCE Repository
First things first, clone the MDANCE repository if you haven’t already.
$ git clone https://github.com/mqcomplab/MDANCE.git
$ cd MDANCE/scripts/prime
2. Cluster Normalization
normalize.py With already clustered data, this script will normalize the trajectory data between \([0,1]\) using the Min-Max Normalization.
# System info - EDIT THESE
input_top = '../../examples/md/aligned_tau.pdb'
unnormed_cluster_dir = '../outputs/labels_*'
output_dir = 'normed_clusters'
output_base_name = 'normed_clusttraj'
atomSelection = 'resid 3 to 12 and name N CA C O H'
n_clusters = 6
Inputs
System info
input_top is the topology file used in the clustering.unnormed_cluster_dir is the directory where the clustering files are located from step 3.output_dir is the directory where the normalized clustering files will be saved.output_base_name is the base name for the output files.atomSelection is the atom selection used in the clustering.n_clusters is the number of clusters used in the PRIME. If number less than total number of cluster, it will take top n number of clusters.Execution
Make sure your pwd is $PATH/MDANCE/scripts/prime.
$ python normalize.py
Outputs
normed_clusttraj.c*.npy files, normalized clustering files.normed_data.npy, appended all normed files together.3. Similarity Calculations
prime_sim generates a similarity dictionary from running PRIME.
-h Help with the argument options.-m Methods, {pairwise, union, medoid, outlier} (required).-n Number of clusters (required).-i Similarity index, {RR or SM} (required).-t Fraction of outliers to trim in decimals (default is None).-w Weighing clusters by frames it contains (default is True).-d Directory where the normed_clusttraj.c*.npy files are located (required)-s Location where summary file is located with population of each cluster (required)Execution
Make sure your pwd is $PATH/MDANCE/scripts/prime.
$ prime_sim -m union -n 6 -i SM -t 0.1 -d normed_clusters -s ../nani/outputs/summary_6.csv
To generate a similarity dictionary using data in
normed_clusters (make sure you
are in the prime directory) using the union method (2.2 in Fig 2) and
Sokal Michener index. In addition, 10% of the outliers were trimmed.
Outputs
w_union_SM_t10.txt file with the similarity dictionary.{
"frame_0": [
0.7, # cluster 1 similarity.
0.9, # cluster 2 similarity.
...,
0.8 # average similarity of all above similarities.
]
}
4. Representative Frames
prime_rep will determine the native structure of the protein using
the similarity dictionary generated in step 5.
-h for help with the argument options.-m methods (for one method, None for all methods).-s folder to access for w_union_SM_t10.txt file.-i similarity index (required)-t Fraction of outliers to trim in decimals (default is None).-d directory where the normed_clusttraj.c* files are located (required if method is None)Execution
Make sure your pwd is $PATH/MDANCE/scripts/prime.
$ prime_rep -m union -s outputs -d normed_clusters -t 0.1 -i SM
Outputs
w_rep_SM_t10_union.txt file with the representative frames index.
Further Reading
For more information on the PRIME algorithm, please refer to the PRIME paper.
Please Cite
@article{chen_protein_2024,
title = {Protein Retrieval via Integrative Molecular Ensembles (PRIME) through Extended Similarity Indices},
issn = {1549-9618},
url = {https://doi.org/10.1021/acs.jctc.4c00362},
doi = {10.1021/acs.jctc.4c00362},
journal = {Journal of Chemical Theory and Computation},
author = {Chen, Lexin and Mondal, Arup and Perez, Alberto and Miranda-Quintana, Ramón Alain},
month = jul,
year = {2024},
note = {Publisher: American Chemical Society},
}
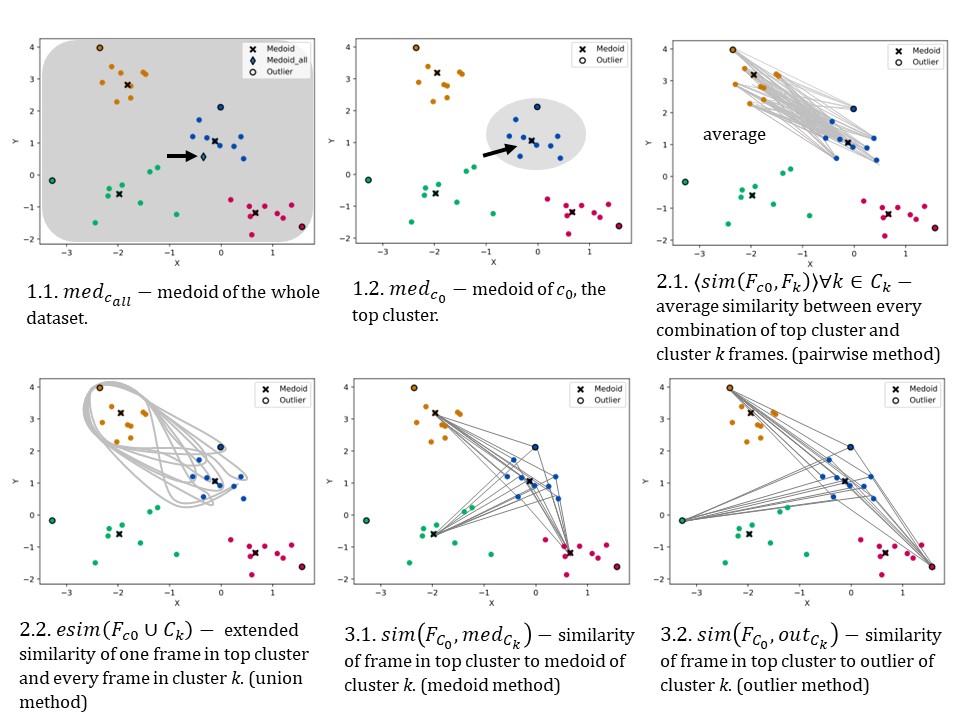
Fig 2. Six techniques of protein refinement. Blue is top cluster.